
Accepted 17 June 2021
Available Online 28 June 2021
- DOI
- https://doi.org/10.2991/mathi.k.210617.001
- Keywords
- Cancer
immune checkpoint blockade
immunotherapy
resistance
bispecific antibodies - Abstract
Cancer is a devastating disease that can be challenging to treat. Fortunately, immune cells called T-cells can be activated for tumour destruction. Signal 1 of T-cell activation is provided by antigen presentation through the (T-cell receptor/major histocompatibility complex) interaction. Signal 2 of T-cell activation is provided by a costimulatory interaction (CD28/CD80). Immune checkpoint blockade (programmed cell death-1/programmed cell death ligand-1 [PD-L1]) is a regulatory interaction controlling T-cell activation/repression. However, tumour cells may exploit checkpoint blockade for tumour survival. Immunotherapy is a favourable cancer therapy provided by ‘the magic bullet’ approach. Monoclonal antibodies provide cytostatic effects against checkpoint blockade. However, tumours may evolve modifications, including tumour antigen loss and tumour ligand overexpression (PD-L1). This can inactivate T-cells, causing immunotherapy resistance and low clinical responses. Bispecific antibodies possess superior properties to monoclonal antibodies provided by their spatio-temporal effects, with potential to improve anti-tumour activity. Bispecific antibodies in development were reviewed for their structures, development technologies and anti-tumour activities. FS118 and M7824 are used to treat refractory cancers, whilst Tebotelimab and XmAb20717 solely enhance T-cell activation for tumour destruction. As cellular factors can switch off T-cell activity leading to immunotherapy resistance, bispecifics with focus on T-cell activity may be ineffective in tumour targeting. This may provide an opportunity to develop a bispecific antibody as part of future research, with binding targets, PD-L1 and lymphocyte-activation gene 3. This may promote cytostatic effects for T-cell activation. Furthermore, effector cell binding may use antibody-dependent cellular cytotoxicity effects to ensure tumour destruction.
- Copyright
- © 2021 The Author. Published by Atlantis Press B.V.
- Open Access
- This is an open access article distributed under the CC BY-NC 4.0 license (http://creativecommons.org/licenses/by-nc/4.0/).
1. INTRODUCTION
1.1. Cancer – A Disease of Cells
Cancer is a devastating collection of diseases related to ageing, affecting one in two people across the UK [1]. Cells in the body begin to grow uncontrollably into a highly proliferative tissue, known as a tumour [2]. These malignant cells use all resources necessary for survival, whilst limiting resources to healthy cells, leading to organ failure and death [3,4]. Due to the increasing prevalence of this malignant disease, much focus in research is directed towards immunotherapy [5]. Intra-tumour heterogeneity describes morphological differences occurring within tumour cells, including gene expression, genomic alterations, and metabolism [6–8]. These cellular changes drive tumour progression and are considered the cause of immunotherapy resistance and treatment failure [9]. Bispecific antibodies are of increasing interest provided by their multiple binding approach [10]. This review therefore aims to familiarize the reader with bispecific antibody structure in relation to their anti-tumour functions.
1.2. ‘The Magic Bullet’
Chemotherapy destroys both malignant and healthy cells [11], whereas immunotherapy offers a targeted approach to stimulate the immune system to destroy malignant cells. The concept of using antibodies to enhance immune responses was first perceived by the German Chemist, Paul Ehrlich, and subsequently developed into ‘the magic bullet’ [12]. Immunotherapy aims to exploit the immune system’s ability to recognise abnormal cells through a specific antigen and destroy them [13]. Antibodies are biological molecules produced by the immune system to respond to antigens, some of which are pathogens which have the potential to cause disease [14]. To protect the body, antibodies can destroy pathogens through cytostatic effects, in which target antigen binding can block signalling pathways. Furthermore, cytotoxic effects provided by Antibody-dependent Cellular Cytotoxicity (ADCC) and complement-mediated cytotoxicity can destroy pathogens [15,16]. As cancer is a continually evolving disease, cancer survival changes can be identified through antigens and targeted by an antibody, ‘the magic bullet’ to effectively destroy the cancer [17].
1.3. The Immune System and Immunotherapy
T-cells play a key role in the adaptive immune system to protect against foreign antigens [18]. T-cells can recognise cancer cells manifested with pathogens and destroy them. Thus, immunotherapy has aimed to enhance T-cell activity against malignant cells [19]. Three signals are required for T-cell activation [17]. Firstly, newly produced T-cells (naïve T-cells) cannot recognise pathogens. Therefore, other cells known as Antigen-presenting Cells (APCs), such as dendritic cells can present a specific antigen to the T-cell Receptor (TCR) using a Major Histocompatibility Complex-II (MHC-II) molecule. Consequently, upon T-cell entry into a Tumour Microenvironment (TME), the T-cell can recognise a tumour cell through the same antigen. The TCR/MHC-II interaction therefore provides signal 1 of T-cell activation [17,20,21]. Secondly, costimulatory receptors, known as immune checkpoint molecules function in further promoting T-cell activity. For instance, CD28 is a stimulatory checkpoint from a T-cell that can bind the ligands, CD80 or CD86 on a dendritic cell, which provides signal 2 of T-cell activation [17,22]. Thirdly, the inflammatory cytokines; Interferons (IFN) and Interleukins (IL) contribute to T-cell proliferation and clonal expansion, which is signal 3 of T-cell activation [17,23,24]. Additional immune checkpoint molecules localised on T-cells may also associate with tumour ligands which prevent T-cell activation. This is known as an immune checkpoint blockade [25]. For example, Programmed Cell Death-1 (PD-1) is a checkpoint inhibitor present on a T-cell which binds to the checkpoint inhibitors, Programmed Cell Death Ligand-1 or 2 (PD-L1 or PD-L2) on a tumour cell [26]. This helps balance T-cell activation and repression to provide immune responses when necessary [27]. However, as immune checkpoint blockade blocks T-cell activation [26], tumour cells can enhance this pathway for tumour survival [17].
Checkpoint inhibitors were first discovered in 1910 when healthy blood samples had shown to dissolve cancer cells, as opposed to blood from cancer patients [28]. In 1987, the first checkpoint inhibitor, Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) was discovered [29], and in 1995 its function was discovered in avoiding T-cell activity [30]. Antibody-based medicines have been developed to target these immune checkpoints which led James Allison and Tasuku Honjo to receive the Nobel Prize in 2018 [31]. Antibodies have been engineered to specifically bind a single epitope (target) on an antigen, termed recombinant monoclonal antibodies [32]. They can bind immune checkpoint inhibitors to obstruct immune checkpoint blockade to enable T-cell activation [33]. Examples of clinically used checkpoint inhibitor antibodies include Nivolumab and Pembrolizumab which can bind PD-1, whilst Atezolizumab, Durvalumab and Avelumab can bind to PD-L1, and Ipilimumab can bind to CTLA-4 [34].
Monoclonal antibodies have a modular tertiary structure which enables them to perform their functions. Antibodies are composed of two heavy chains and two light chains joined by disulphide bonds [35]. Two antigen-binding fragments (Fab) in the variable region can bind an antigen (e.g. checkpoint inhibitor) containing either one or two (same) epitopes [33]. Through binding two epitopes this can provide the property of avidity; a strong binding force to enhance antigen targeting [36]. There is also a fragment crystallizable (Fc) fragment important for inducing ADCC activity, along with antibody recycling to increase the half-life [16,37].
1.4. Immunotherapy Resistance
Despite the growing use of immunotherapy to treat immune checkpoint blockade, only a small subset of patients is responsive to this therapeutic approach, including varied patient success rates [38,39]. To promote tumour survival, cancer cells may promote cellular modifications to switch off T-cell activation, thereby promoting resistance to antibody-based therapies [40,41].
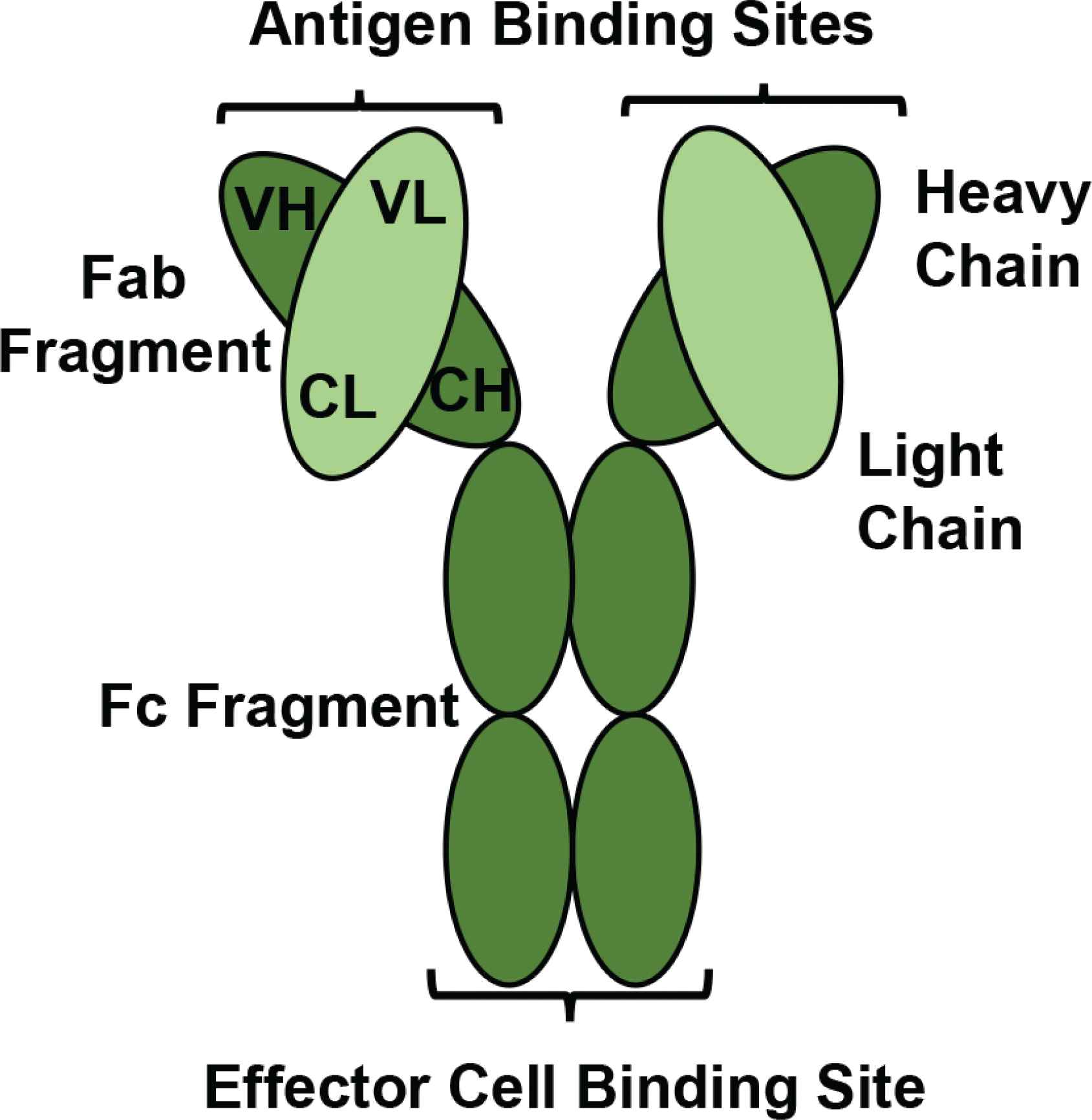
A schematic diagram of a monoclonal antibody. A monoclonal antibody consists of four polypeptide chains; two heavy chains containing Variable Heavy (VH) and Constant Heavy (CH) regions are bound together by two disulphide bonds. The heavy chains are attached to two light chains containing Variable Light (VL) and Constant Light (CL) regions by one disulphide bond. There is an Fc fragment for effector immune cell binding, and two Fab fragments each provide an antigen binding site for epitope recognition. (Figure developed from Moorthy et al. [35])
One reason for developing resistance to T-cell-based therapies is the inability of tumour recognition due to tumour antigen loss [42], or failure to present the antigen [43,44]. The Major Histocompatibility Complex-I (MHC-I) molecule from a tumour cell can present an antigen to a T-cell, allowing tumour cell recognition [17]. However, a mutation may occur in MHC-I, more specifically Beta 2-Microglobulin (B2M), which reduces expression of B2M and impairs expression of MHC-I [43–45]. Consequently, antigens cannot successfully present on the tumour surface, thereby averting the interaction between the immune system and tumour cell for T-cell activation [45,46]. Another reason for immunotherapy resistance is tumour ligand overexpression [47]. The tumour ligand, PD-L1 has been overexpressed in many cancer-cell lines [48]. PD-L1 commonly functions in immune checkpoint blockade to regulate T-cell activity [26], along with intracellular signalling to promote cancer survival [47]. IFNs are signalling proteins produced by T-cells which can upregulate transcriptional activity of PD-L1 [49]. IFNs provide T-cell cytotoxicity against cancer cells, however, cancer cells can evade this T-cell attack through upregulating PD-L1 to exploit the PD-1/PD-L1 immune checkpoint blockade. There is also release of intracellular anti-apoptotic signals by PD-L1 [50]. A study has shown that PD-L1 contains the intracellular signalling motifs, RMLDVEKC and DTSSK which provide cancer protection against apoptotic activities provided by IFN [50].
1.5. Bispecific Antibody-Based Medicines
With the greater shift toward immunotherapy to treat malignant diseases, efforts continue to develop antibodies with improved clinical outcomes [51]. A bispecific antibody binds two antigens or two different epitopes of an antigen [10]. They provide superior properties to monoclonal antibodies through providing spatio-temporal effects for improved target binding and enhanced immune responses [10].
T-cell redirectors are the most advanced bispecific antibodies having reached two biologics to the clinic. Blinatumomab (Blincyto) is a bispecific T-cell engager [52] and Catumaxomab (Removab) is a trifunctional bispecific antibody. There are also dual immunomodulatory bispecific antibodies in clinical development, which bind two immunomodulating targets (immune checkpoint molecules) [10]. Combination therapy employs more than one monoclonal antibody (one binding target) for cancer therapy [53], whereas bispecific antibodies have the benefit of binding two targets. Bispecifics can therefore provide spatio-temporal binding effects, including greater binding specificity to two antigens simultaneously or sequentially [54]. Bispecifics also avoid potential dose toxicity which is commonly observed in combination therapies [53]. Examples of dual immunomodulators in clinical development are shown below.
2. FS118: PD-L1 × LAG-3
2.1. Bispecific Design and Structure
FS118 is a bispecific antibody developed by the company F-star to bind PD-L1 and Lymphocyte-activation Gene 3 (LAG-3) antigens [55]. MHC-II from a dendritic cell presents an antigen to the TCR of a T-cell [17,20,21]. However, an MHC-II/LAG-3 interaction can cause continuous antigen stimulation and exhausted T-cell function [25], thereby supporting LAG-3 as an FS118 target [55]. Furthermore, as PD-L1 can bind to PD-1 causing immune checkpoint blockade [26], this supports PD-L1 as a viable target by FS118 [55]. F-star has engineered FS118 to have PD-L1 binding via the variable region and LAG-3 binding through the constant region. Thus, allowing targeting through opposing structural ends of the antibody, with the aim of treating resistant and refractory cancers [55], see Figure 2.
2.2. Anti-tumour Function
To investigate target binding of FS118, a Surface Plasmon Resonance (SPR) assay was undertaken [61]. This procedure involved one ligand (e.g. PD-L1) attached to the surface of a sensor chip, while the binding molecule FS118 was passed over the surface [62]. A response was generated according to extent of binding. A reaction favouring association is defined as the equilibrium association constant (ka), whilst dissociation is defined by the equilibrium dissociation constant (kd). The ratio of kd/ka is known as the equilibrium dissociation constant kD, which is when rate of association and dissociation are in equilibrium. Small kD values are known to produce high binding affinities, which is ideal for effective target binding [63]. FS118 showed greater association (6.30) than dissociation (3.50) to LAG-3, reflecting a small kD value of 0.06 nM. In comparison, binding to PD-L1 showed greater dissociation (3.90) than association (0.38), resulting in a larger KD value of 1.00 nM. This has indicated that FS118 has high affinity and potent binding to LAG-3, with moderate affinity to PD-L1 [61].
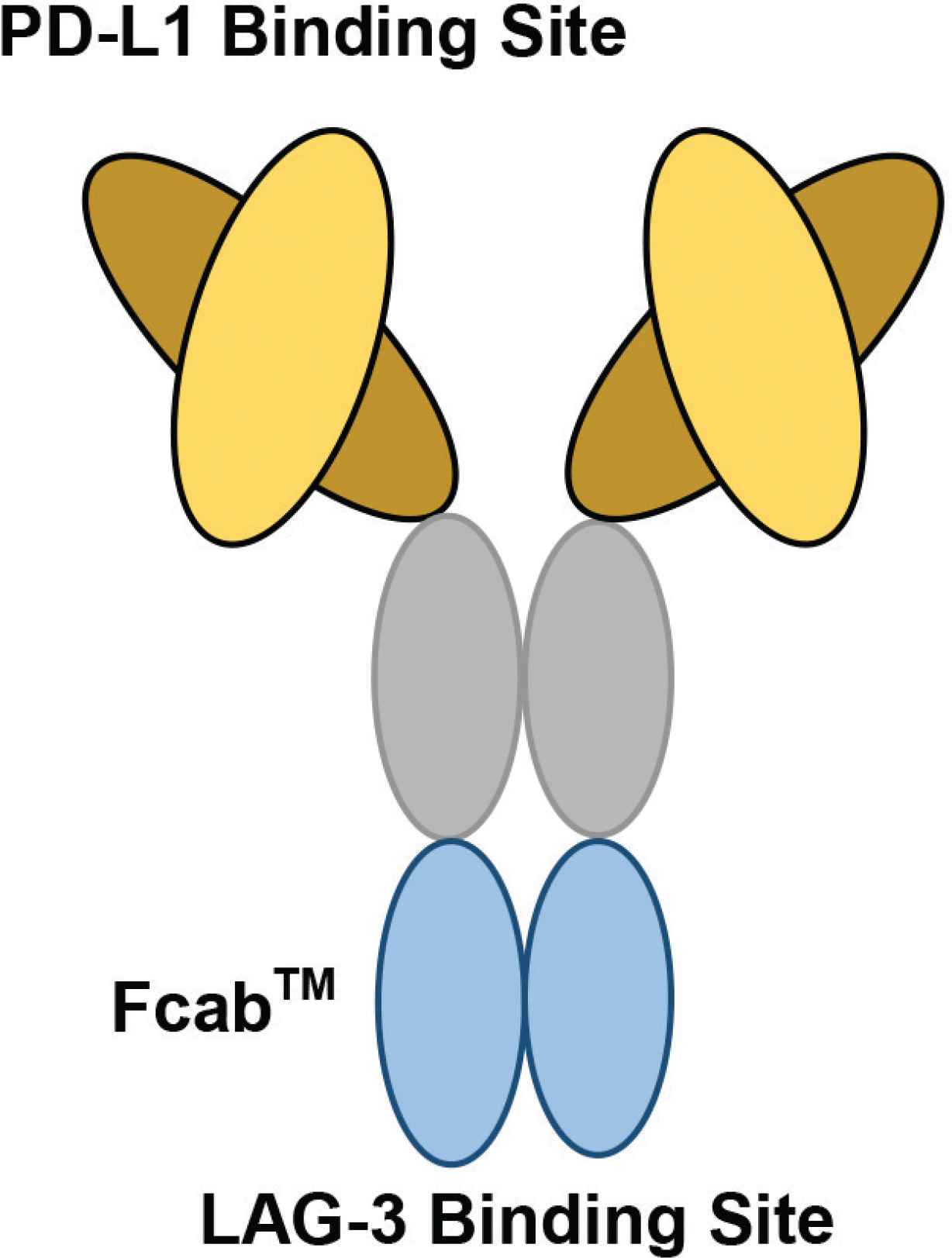
The structure of F-star’s bispecific antibody, FS118. F-star has used a technology platform to create a novel antigen binding site at the Fc region, known as Fcab™ (Fc-domain with antigen-binding activity). Fcab is joined to the Fab regions to produce a full-length Immunoglobulin (IgG) antibody, known as a tetravalent mAb2 bispecific. On FS118, PD-L1 binding occurs at the variable region, whilst LAG-3 binding occurs at the constant region (Figure redrawn from Kraman et al. [59] and F-star Therapeutics [60]).
| Bispecific antibodies | Target antigens | Structural format/technology | Clinical phase and identifier | References |
|---|---|---|---|---|
| FS118 | PD-L1 × LAG-3 | mAb2 bispecific antibody | Phase I/II NCT03440437 | [55] |
| M7824 | PD-L1 × TGF-β | Bifunctional fusion protein | Phase I/II NCT04297748 | [56] |
| Tebotelimab (MGD013) | PD-1 × LAG-3 | Tetravalent DART® molecule | Phase I/II NCT04212221 | [57] |
| XmAb20717 | PD-1 × CTLA-4 | XmAb® Technology | Phase I NCT03517488 | [58] |
Immunomodulatory bispecific antibodies in clinical development. Bispecific antibodies have been researched for their immune checkpoint targets; common targets include PD-1, PD-L1 and LAG-3. Each bispecific has been developed with a unique technology to provide an overall structural format with bispecific properties. The Clinicaltrials.gov identifier has also been provided to exhibit clinical testing
Upon T-cell activation, ILs are produced and provide the third signal for T-cell clonal expansion [17,23,24]. Therefore, ligand binding and T-cell activation by FS118 are reflected by the amount of IL-2 production. Monoclonal antibodies and mAb2 antibodies were incubated with DO11.0 cells (overexpressing LAG-3) and LK35.2 cells (overexpressing PD-L1) for 2 h. IL-2 concentrations were then measured by Enzyme-linked Immunosorbent Assay (ELISA) as a marker of T-cell activation [61]. The highest IL-2 production was shown by a combination of antibodies binding to LAG-3 and PD-L1 individually, followed by FS118 and then a combination treatment of an mAb2 LAG-3 antibody and individual PD-L1 antibody. However, treatments including a single antibody target showed very low IL-2 production, suggesting fewer activated T-cells [61]. Despite FS118 not having the highest IL-2 concentration compared to the combination treatments, FS118 may still activate T-cells, reflected by a high level of IL-2 production. Furthermore, FS118 combines targeting of both ligands in one bispecific antibody. This has demonstrated the potential of FS118 in replacing combination therapy for effective T-cell activation [61].
To examine anti-tumour activity of FS118 in tumour-bearing mice, tumour cells from the MC38 cancer cell line were injected into C57/Bl6 mice to develop palpable tumours. The mice were then administered three doses of antibody treatments on days 8, 11 and 14 to examine a decrease in tumour volume [64]. Mice administered FS118 had the smallest tumour volumes, followed by those administered the combination treatment (LAG-3 mAb + PD-L1 mAb). However, mice given treatments including one antibody target showed larger tumour volumes. Furthermore, the IgG antibody (control) was not sufficient to produce anti-tumour responses, therefore the tumours could progress in these mice. From the mice given FS118, 6/8 mice were tumour-free after 20 days. However, tumour volume may not correlate with overall survival. For instance, combination therapy showed direct tumour regression but none of the seven mice had survived after 20 days [64].
2.3. Immune Checkpoint Targets
FS118 was designed to target the immune checkpoints, PD-L1 and LAG-3, and treat advanced tumours and Head and Neck Squamous Cell Carcinoma [65]. FS118 has shown to provide moderate binding to PD-L1 [61], which has been overexpressed in many cancer-cell lines [48]. Therefore PD-L1 is a favourable antibody target in avoiding checkpoint blockade [26,55]. FS118 has mainly been developed to target upregulated LAG-3. LAG-3 causes T-cell exhaustion and FS118 has shown strong binding to LAG-3 [61]. FS118 can bind to both a tumour cell (PD-L1) and T-cell (LAG-3) bringing both cells in close vicinity to block immune checkpoint blockade and prevent T-cell exhaustion. A potential weakness of FS118 may be the inability to use the dual binding approach simultaneously due to the differences in binding affinity. This could lead to LAG-3 binding but no PD-L1 binding, resulting in a reduced anti-tumour effect.
3. M7824: PD-L1 × TGF-β
3.1. Bispecific Design and Structure
M7824 is a recombinant bifunctional fusion protein engineered to bind simultaneously to the ligands, PD-L1 and Transforming Growth Factor-Beta (TGF-β) [56,66]. M7824 was developed by the companies Merck and GSK to treat refractory cancers to avert development of cancer resistance [67]. The variable region binds to PD-L1, whilst the constant region is composed of two TGF-β Receptor (TGF-βR) molecules, which serve as a trap for TGF-β [67]. TGF-β is a regulatory cytokine involved in cell development and maintenance with high expression in the TME [68–70]. Upon TGF-βR binding, a signal is released to induce tumour progression through angiogenesis [71], contribute to the Epithelial Mesenchymal Transition (EMT) [72] and inhibit immune functions [73,74]. M7824 can therefore prevent intrinsic and extrinsic evasion mechanisms to stop tumour progression.
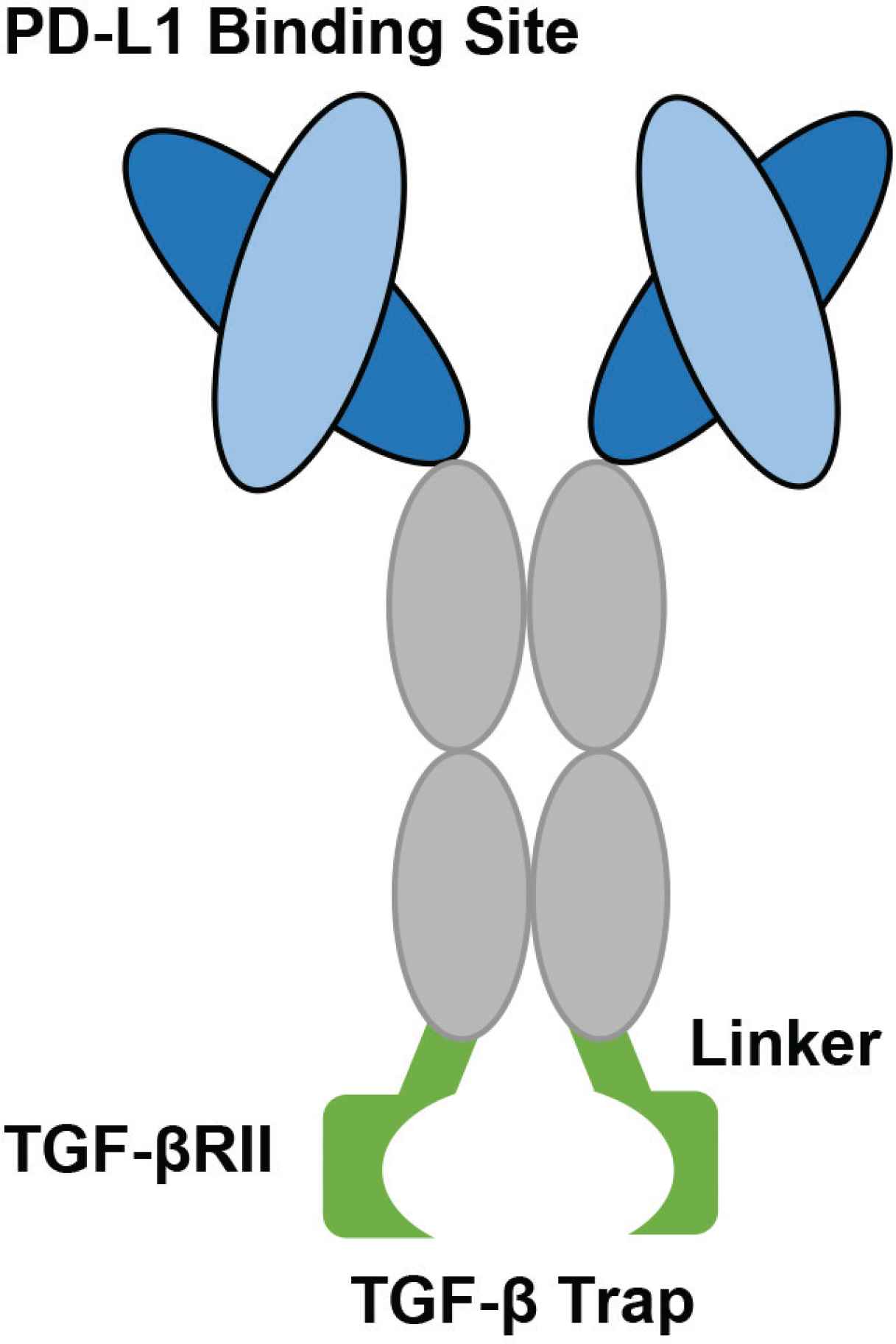
The structure of the bifunctional fusion protein, M7824. The light and heavy chains are identical to the monoclonal antibody, Avelumab. The constant region contains three amino acid substitutions fused to the N terminus of two TGF-β RII molecules using recombinant (Gly4Ser)4Gly linkers. The two TGF-β RII molecules can bind and trap a TGF-β molecule (Figure developed from Lan et al. [67]).
3.2. Anti-tumour Function
The binding of M7824 to the ligands, PD-L1 and TGF-β was analysed both individually and concomitantly. M7824 binding to cells overexpressing PD-L1 from the Human Embryonic Kidney (HEK) cell line was assessed by flow cytometry, through the Mean Fluorescence Intensity (MFI). M7824 binding to TGF-β1, TGF-β2 and TGF-β3 was analysed by ELISA, and simultaneous binding was assessed using horseradish peroxidase conjugated to streptavidin through the Optical Density (OD) [67]. Both M7824 and an anti-PD-L1 molecule showed effective binding to PD-L1, reflected by high MFI values, compared to trap control with hardly any binding to PD-L1. M7824 showed effective binding to TGF-β1, less effective binding to TGF-β3 but no binding to TGF-β2, whilst anti-PD-L1 showed no binding to TGF-β [67]. Binding to TGF-β2 was further assessed and M7824 showed sufficient binding affinity to TGF-β2. M7824 also demonstrated simultaneous binding reflected by a high OD 450 nm value [67]. These results have indicated that M7824 has effective individual and simultaneous binding to PD-L1 and TGF-β. The reason that M7824 first showed no binding to TGF-β2, whereas a separate investigation showed efficient binding is due to low intrinsic binding between plate-bound TGF-β2 and the TGF-βRII [75]. However, TGF-β molecule in solution facilitated a strong binding avidity between TGF-β2 and the receptor due to bivalency [67]. These results are encouraging and advantageous for M7824, as TGF-β can be transported around the body through bodily fluids, and M7824 can provide adequate target binding in vivo [76].
T-cell activation of M7824 was then assessed in vitro. Serial dilutions containing M7824 were incubated with Human Peripheral Blood Mononuclear Cells (huPBMCs) and IL-2 production was examined using ELISA. The effect of M7824 on TGF-β signalling was also examined using a luciferase assay [67]. Cells administered M7824 showed an instant increase in IL-2 production at roughly 100 ng/ml of M7824. IL-2 production was then maintained with increasing M7824 concentration. M7824 showed a sudden decrease in TGF-β signalling at approximately 50 ng/ml of M7824, and low signalling was also maintained with increasing M7824 concentration, whereas anti-PD-L1 showed no change [67]. These results have demonstrated that PD-L1 binding from M7824 facilitated T-cell activation, reflected by the increase in IL-2 production, whilst the TGF-β trap method helped to reduce tumour signalling processes.
The anti-tumour responses provided by M7824 was analysed in mice inoculated with cells from the EMT-6 or MC38 cancer cell lines. This allowed tumour development in the mice and the tumour sizes were investigated following treatment administration [67]. The mice inoculated with either the EMT-6 or MC38 cell lines and given M7824 (164 and 492 µg) had the smallest tumour volumes. Mice given the combination treatment of anti-PD-L1+ trap control (133 and 164 µg) showed larger tumour volumes with comparable results. Mice given anti-PD-L1 (133 and 400 µg) had larger tumour volumes, whilst mice given trap control and isotype control showed the largest tumour volumes [67]. Both M7824 and combination treatments have two binding targets and have shown the smallest tumour volumes in the mouse subjects, indicating superior drug efficacy. However, the treatments binding to one target have shown larger tumour volumes in mice, indicating reduced anti-tumour efficacy.
3.3. Immune Checkpoint Targets
M7824 was developed to prevent development of resistant cancers associated with the PD-1/PD-L1 blockade and TGF-β signalling [67]. Therefore, targeting PD-L1 can prevent immune checkpoint inhibition, while targeting TGF-β can decrease tumour signalling processes. When analysing binding of M7824, there was effective PD-L1 binding via the two Fab fragments which may indicate bivalency. The anti-PD-L1 moieties correspond to those from the monoclonal antibody, Avelumab [67], which is known to exhibit bivalency [77]. This could suggest that M7824 may also exploit bivalency for PD-L1, though there was insufficient data to support this. Despite this, as the constant region of M7824 was composed of two TGFβRII molecules, M7824 could exploit bivalency for TGF-β [67]. This may indicate M7824 having bivalency for its targets via both its variable and constant regions, thereby reflecting effective immune checkpoint target binding.
4. TEBOTELIMAB: PD-1 × LAG-3
4.1. Bispecific Design and Structure
Tebotelimab is an IgG4 bispecific antibody developed by MacroGenics to bind the checkpoints, PD-1 and LAG-3 located on exhausted T-cells [57]. This allows blocking of inhibitory interactions with other molecules, including PD-L1, PD-L2 and MHC-II. Tebotelimab maintains a tetravalent structure generated using a Dual Affinity Re-Targeting (DART®) platform [78]. Regular bispecific antibodies contain two Variable Heavy (VH) chains joined at the hinge region, though reducing structural flexibility upon antibody-antigen recognition. Fortunately, the DART® technology can address this conformational constraint to improve the binding efficiency [79,80].
4.2. Anti-tumour Function
Tebotelimab binding to PD-1 and LAG-3 was assessed in vitro by Fluorescence-activated Cell Sorting (FACS) [81]. Cells from the NS0 cell line were engineered to contain PD-1 or LAG-3. Upon Tebotelimab and comparative antibody administration, target binding can be reflected by an increase in the MFI [81]. There was effective binding to PD-1+ NS0 cells by Tebotelimab and marketed Nivolumab, reflected by an increase in the MFI, with no binding by marketed 25F7 (anti-LAG-3). There was active binding to LAG-3 by both Tebotelimab and 25F7, shown by an increase in the MFI, and no binding by Nivolumab which only binds to PD-1 [81]. This data has indicated that Tebotelimab has efficient target binding and is comparable to antibodies on the market, suggesting the immunotherapy as a promising therapeutic.
Interferons are generated by T-cells to provide T-cell cytotoxicity for tumour cell destruction. IFNγ induction by Tebotelimab was therefore assessed through a Staphylococcus Enterotoxin B stimulation assay as a marker of T-cell activation [81]. Tebotelimab showed the highest IFNγ production, which indicates its potential in promoting sufficient T-cell cytotoxicity. This bispecific is also superior to the combination of MacroGenics anti-PD-1 (MGDA012) and Macrogenics anti-LAG-3, and the combination of marketed Nivolumab and 25F7* [81]. This has implied that Tebotelimab would be far more effective than using combination therapy in promoting T-cell activation to destroy cancer cells. Furthermore, antibodies with one binding target; including MGDA012, Nivolumab, Macrogenics anti-LAG-3 and 25F7 showed low IFNγ induction, which may result in fewer activated T-cells.
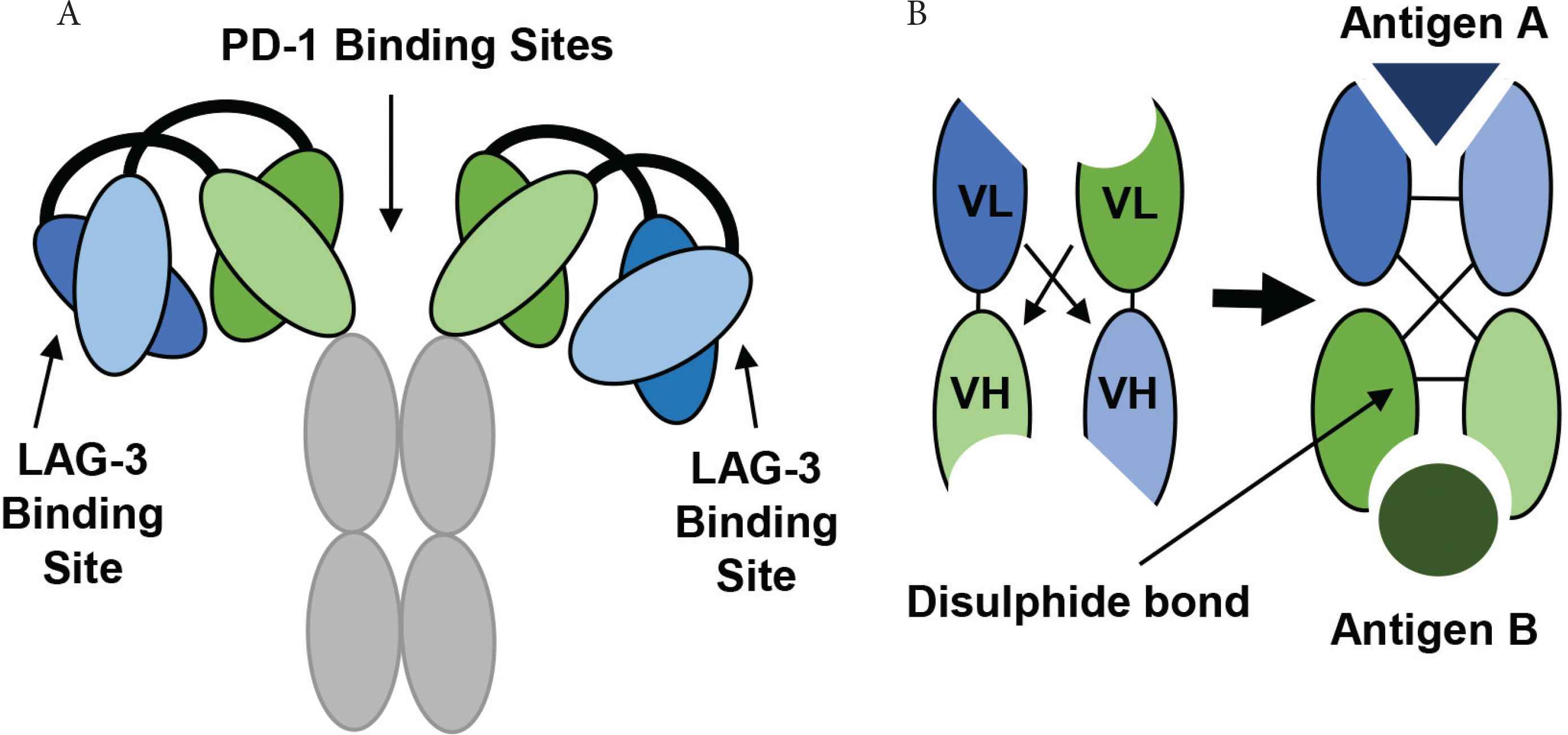
The development and structure of Tebotelimab using DART® technology. (A) The tetravalent DART molecule, Tebotelimab can bind two PD-1 and two LAG-3 molecules. The variable chains from two monoclonal antibodies combine through a disulfide linkage to form a bispecific DART molecule with two binding targets. (B) The VL and VH chains from one antibody join to produce the first antigen binding site, while the VL and VH chains from another antibody produce a second antigen binding site (Figure redrawn from Creative Biolabs [79] and MacroGenics [80]).
Anti-tumour activity of Tebotelimab was analysed from patients with Triple-Negative Breast Cancer (TNBC), Epithelial Ovarian Cancer (EOC) and Non-Small Cell Lung Cancer (NSCLC) in the first in-human clinical trial. Anti-tumour activity was investigated through observing a change in target lesions [82].
There were 23 evaluable patients with TNBC, of which more than 50% (60.9%) had disease progression, indicating tumour growth and less effective treatment. Furthermore, 34.8% have shown stable disease with very few patients showing treatment responses (4.3%). There were also 23 patients with EOC, with less than 50% with disease progression (47.8%). In addition, less than 50% of patients (43.5%) showed stable disease, thereby indicating insignificant tumour growth for these patients. Few patients also showed to respond well to treatment (8.7%). From the NSCLC CPI-naïve patients, 50% of patients showed stable disease, with fewer patients with disease progression (35.7%) and some patients responded to treatment (14.3%). From all cancer patients, those with NSCLC post-PD-1 appeared to have the most patients with stable disease (53.3%), with slightly fewer patients with disease progression (46.7%). However, none of the patients showed partial response (0%), suggesting limitations in providing clinical responses [82]. The results above have indicated that few cancer patients show clinical responses to Tebotelimab, though most patients present with stable disease or disease progression. This has suggested that Tebotelimab presents with limitations and requires further research to be considered as an effective immunotherapy for clinical use.
4.3. Development Technology
MacroGenics had focused on the variable region of Tebotelimab, using DART technology to improve target binding efficiency [78,79]. There was effective binding of Tebotelimab to both PD-1 and LAG-3 [81]. As target binding blocks negative interactions to enable T-cell activation, this was analysed through the extent of IFNγ induction as a clinical marker. There was a great increase in IFNγ production upon Tebotelimab administration, thereby indicating T-cell activation [81]. Another example of DART technology was used to develop the bispecific, Flotetuzumab. Flotetuzumab binds to a T-cell (CD3) and tumour cell (CD123) and is known as a T-cell redirector [83]. This antibody has shown effective target binding through a BIAcore SPR assay, resulting in an adequate cytolytic potency provided by CD3. Anti-tumour activity was also demonstrated favourably in tumour-bearing NOD scid gamma (NSG) mice [84]. This has indicated that DART technology can generate bispecific structures with effective target binding functions. Anti-tumour activity provided by Tebotelimab was assessed in a human clinical study [82]. Few patients showed clinical responses reflected by tumour reduction, whilst most patients presented with stable and progressive diseases [82]. As Tebotelimab was used to treat unresectable and metastatic neoplasms, the treatment may prevent disease progression for some patients. However, Tebotelimab alone was unable to sufficiently eradicate the tumour. The clinical trial also investigated the combination with Margetuximab (anti-HER2 antibody) [82]. Margetuximab contains an Fc region modified by the MacroGenics Fc Optimization platform for effector cell binding [85]. This treatment showed enhanced anti-tumour responses in relapsed/refractory HER2-positive solid tumours with an objective response rate of 42.9% [81]. Despite the improved clinical responses, this may be predominantly due to Margetuximab as opposed to Tebotelimab.
| TNBC | EOC | NSCLC, CPI-Naïve | NSCLC, post-PD-1 | |
|---|---|---|---|---|
| Number of patients | 23 | 23 | 14 | 15 |
| ORR (Confirmed) | 4.3% | 8.7% | 14.3% | 0% |
| ORR (Confirmed + Unconfirmed) | 17.4% | 8.7% | 21.4% | 13.3% |
| Stable disease | 34.8% | 43.5% | 50.0% | 53.3% |
| Disease progression | 60.9% | 47.8% | 35.7% | 46.7% |
Data obtained for anti-tumour activity following Tebotelimab administration. The proportion of cancer patients with Objective Response Rate (ORR), stable disease, and disease progression, following Tebotelimab administration (Table developed from Luke et al. [82])
5. XMAB20717: PD-1 × CTLA-4
5.1. Bispecific Design and Structure
XmAb20717 is a unique bispecific antibody designed by Xencor to target PD-1 and CTLA-4 checkpoint molecules. This prevents interactions with PD-L1, PD-L2 and CD80 with the aim of activating T-cells. XmAb20717 was engineered using an XmAb® platform to provide IgG-like antibody properties, whilst altering the Fc region [58]. When an antibody binds its target, this can trigger Fc-Gamma Receptor (FcγR) binding to an effector cell, e.g. Natural Killer (NK) cell for ADCC functions [86]. However, as XmAb20717 targets a T-cell, ADCC activity would lead to T-cell depletion. It therefore contains a modified Fc region to avoid effector cell binding [58]. Moreover, the Fc region can provide an increased antibody circulation time through binding the Neonatal Fc Receptor (FcRn) for recycling [37]. Despite this, antibodies may still possess low circulating half-lives. Xencor has therefore altered the Fc region using Xtend® technology to further enhance the half-life. Two amino acid substitutions provide a threefold increased binding affinity to FcRn. This allows less frequent dosing, greater drug efficacy and reduced manufacturing costs [87,88].
5.2. Anti-tumour Function
XmAb20717 was tested for its target binding ability in huPBMCs from multiple healthy donors which were engrafted into NSG mice [90]. These studies were undertaken to understand T-cell activation, reflected by IL-2 production, CD45+ cell count and IFNγ production.
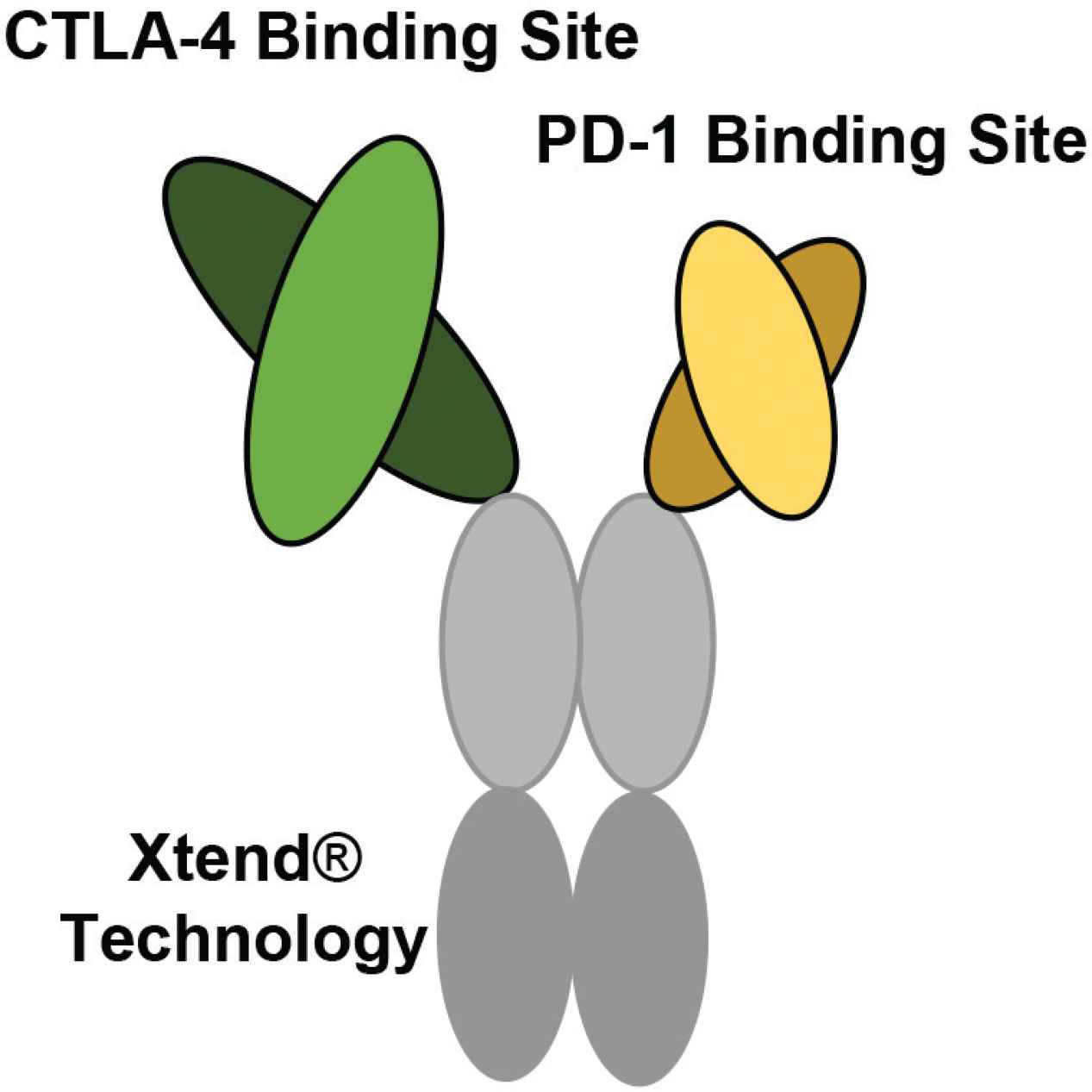
The bispecific structure of XmAb20717 developed by Xencor. XmAb20717 is generated using XmAb® technology. One regular size Fab fragment (arm) targets CTLA-4, and a shorter fragment (arm) targets PD-1. This bispecific also contains an Fc region altered by Xtend® technology to increase the circulating half-life (Figure developed from Hedvat et al. [89]).
XmAb20717 target binding was compared to one-arm antibodies which can bind a single target. To ensure T-cell activation, an antibody must adequately bind its targets. The one-arm antibodies have a certain affinity for binding PD-1 and CTLA-4. In combination therapy, this showed T-cell activation, reflected by an increase in IL-2 and IFNγ production and CD45+ count. However, XmAb20717 has the benefit of binding two antigens (PD-1 and CTLA-4) which can exploit the property of avidity. This therefore provided a stronger target binding force, shown by the superior increase in IL-2 and IFNγ production and CD45+ count [90].
Another study compared the impact of binding avidity of XmAb20717 and anti-PD-1 and CTLA-4 bivalent antibodies [90]. The anti-PD-1 bivalent showed the smallest increase in IL-2 and IFNγ production and CD45+ count. This may reflect a monoclonal antibody binding to PD-1 that can exploit avidity through bivalent target binding. However, XmAb20717 showed superior binding avidity to the anti-PD-1 bivalent, reflected by higher IL-2 and IFNγ production and CD45+ count. XmAb20717 can bind both PD-1 and CTLA-4 also with a strong binding avidity, suggesting enhanced cytostatic effects to obstruct immune checkpoint blockade, and allow T-cell activation more effectively. The anti-PD-1 and anti-CTLA-4 bivalents in combination have shown similar results to XmAb20717 [90]. The reason for this is they can bind the same two targets with strong binding avidities, facilitating cytostatic effects for PD-1 and CTLA-4 just as effectively. The bivalents were used in combination, whereas the comparable XmAb20717 support its use clinically in potentially replacing combination therapy.
Anti-tumour responses in mice were assessed to determine the overall efficacy of XmAb20717. Three groups of NSG (immunodeficient) mice were administered different treatments, along with huPBMCs (containing T-cells and B-cells). Group 1 mice were administered no huPBMCs as a control variable, group 2 mice were administered huPBMCs and group 3 mice were administered huPBMCs with XmAb20717 [90]. Group 1 mice with no huPBMC engraftment had the greatest tumour burden after 21 days due to the absence of an immune system. Group 2 mice with huPBMC engraftment had a lower tumour burden, provided by some immune responses, with few subjects showing variation. Group 3 mice who were administered huPBMC and XmAb20717 had the greatest decrease in tumour burden. There was an enhanced immune response as XmAb20717 can bind its targets to promote T-cell activation, thus improving the treatment outcome; subjects also showed variation [90].
5.3. Development Technologies
Xencor had focused on the Fc ‘stem’ of XmAb20717 to improve clinical responses [58]. XmAb20717 was engineered using Xtend technology for an increased circulation time [87,88]. XmAb20717 binding was assessed in relation to T-cell activation, which showed a strong binding avidity and comparable binding to bivalent antibodies used in combination, reflecting increased T-cell activity. Anti-tumour activity was also assessed and XmAb20717 had effectively decreased the tumour burden in mice [90]. XmAb22841 is another bispecific (CTLA-4/LAG-3 targets) that possesses Fc modifications by Xtend technology [91,92]. XmAb22814 was used in combination with anti-PD-1 antibodies for ‘Triple Checkpoint Blockade’, which has shown selective target binding, enhanced T-cell activation, and allogenic anti-tumour responses [92]. These results support the use of Xtend technology to facilitate a longer duration of action for improved clinical responses. The Fc domain of XmAb20717 was modified to prevent ADCC responses to avoid T-cell depletion [58]. However, Xencor has also developed antibodies to promote ADCC effector functions using a Cytotoxic Fc Domain Platform. Two amino acid changes in the Fc domain allow a 40-fold higher binding affinity to FcγRIIIa on NK cells [93]. For instance, Elipovimab was developed by Xencor in association with Gilead Sciences, containing an Fc domain with both cytotoxic and Xtend activity [94]. This has indicated the feasibility in developing an antibody, including ADCC functions for anti-tumour activity and a half-life to facilitate longer clinical responses.
5.4. Future Research – Novel Bispecific Antibody
Specialist technologies have been used to develop bispecific antibodies to bear structural formats with functional properties. FS118, M7824, Tebotelimab and XmAb20717 share the same immune checkpoint binding targets, including PD-1 and PD-L1. This blocks immune checkpoint blockade to enforce T-cell activation for anti-tumour responses. However, these bispecifics are T-cell dependent; upon failure to activate T-cell activity due to loss of tumour antigen presentation, there would be no alternative source of tumour destruction. This may provide an opportunity to support a new idea for a bispecific antibody as part of future research. The development technologies, DART and XmAb technologies focused on improving activity of the variable and constant regions, respectively. Therefore, combining these technologies to construct a novel bispecific antibody may employ specialist properties with superior functions. DART technology may provide improved target binding, whilst Xtend technology may increase the antibody’s circulation time. Furthermore, a Cytotoxic Fc Domain Platform may provide ADCC activity from an effector cell for additional tumour cytotoxicity.
XmAb20717 was engineered to evade ADCC functions as this could promote T-cell damage [58]. The Cytotoxic Fc Domain Platform would therefore apply better to a bispecific that binds only a tumour cell or both a T-cell and tumour cell. An antibody binding to a T-cell and tumour cell can guide both cells in close vicinity, potentially facilitating more effective T-cell activation [95,96]. From this review, potential T-cell and tumour cell targets may include PD-1/PD-L1, this would prevent immune checkpoint blockade (one functional activity). Other potential targets may include LAG-3/PD-L1, this can block immune checkpoint blockade and T-cell exhaustion (two functional activities) [25,26]. A bispecific with LAG-3/PD-L1 targets may therefore provide superior anti-tumour activity compared to PD-1/PD-L1 targets. FS118 also contains LAG-3/PD-L1 targets, providing one source of tumour destruction (T-cell activity). Although, the novel bispecific would possess both target binding and effector cell binding, providing two sources of tumour destruction (T-cell activity and ADCC effects). For these reasons, the novel bispecific antibody may provide superior anti-tumour functions and improved clinical outcomes, compared to FS118.
6. CONCLUSION
This review has briefly described how T-cells can recognise tumour cells through the presence of antigens to destroy the malignant cells. Immunotherapy can enhance T-cell activity for superior anti-tumour effects. However, tumour survival factors can emerge causing immunotherapy resistance and tumour progression. Resistance can be caused by loss of tumour antigen presentation, resulting in no T-cell/tumour cell recognition. Resistance can also be caused by tumour ligand overexpression, which enhances immune checkpoint blockade to switch off T-cell activation. Bispecific antibodies are in development to bind immune checkpoint molecules with the aim of improving anti-tumour success rates. In this review, the bispecific antibodies, FS118, M7824, Tebotelimab and XmAb20717 were analysed for their structures, development technologies and anti-tumour functions.
All four bispecifics facilitated effective target binding and T-cell activation provided by their specialist bispecific characteristics. FS118 and M7824 were developed to treat resistant and refractory cancers and have shown effective anti-tumour activities. XmAb20717 aimed to promote T-cell activity and has demonstrated effective anti-tumour functions comparable to combination therapy. However, anti-tumour activity from Tebotelimab has shown only few patient responses suggesting treatment limitations. These bispecifics bind the common targets, PD-1 and PD-L1 to block immune checkpoint blockade and promote T-cell activation. However, the restriction of T-cell activation caused by loss of tumour antigen presentation could lead to tumour progression. The author therefore took this opportunity to propose an idea for an improved antibody-based therapy. A novel bispecific antibody would require effective target binding; therefore, DART technology could be used. Xtend technology was used for XmAb20717 to improve the circulation time. FS118 and M7824 cannot utilize antibody recycling due to the lack of an Fc fragment, and despite the presence of an Fc fragment for Tebotelimab, there was greater focus on the variable region. Xtend technology could be used to develop the improved bispecific with an enhanced antibody half-life. Furthermore, the Cytotoxic Fc Domain Platform could be used by the bispecific to provide ADCC effects as an additional source of tumour destruction. Optimal immune targets were determined as LAG-3 (T-cell) and PD-L1 (tumour cell). T-cell and tumour cell binding may direct both cells in close proximity to better stimulate T-cell activity, while ADCC functions may also effectively destroy the cancerous cells.
CONFLICTS OF INTEREST
The author declares have no conflicts of interest.
ACKNOWLEDGMENTS
The Author would offer special thanks to Professor Steve Brocchini and Dr Sahar Sheikh Hassan Awwad for their support and constructive recommendations on this Review. Also like to thank her Grandfather, Ramesh Virji Chouhan for his endless support and encouragement.
ABBREVIATIONS
- ADCC,
antibody-dependent cellular cytotoxicity;
- APC,
antigen-presenting cell;
- B2M,
beta 2-microglobulin;
- CH,
constant heavy;
- CL,
constant light;
- CTLA-4,
cytotoxic T-lymphocyte antigen 4;
- DART,
dual affinity re-targeting;
- ELISA,
enzyme-linked immunosorbent assay;
- EMT,
epithelial mesenchymal transition;
- EOC,
epithelial ovarian cancer;
- Fab,
antigen-binding fragment;
- FACS,
fluorescence-activated cell sorting;
- Fc,
fragment crystallisable;
- Fcab,
Fc-domain with antigen-binding activity;
- FcRn,
neonatal Fc receptor;
- FcγR,
Fc-gamma receptor;
- HEK,
human embryonic kidney;
- huPBMCs,
human peripheral blood mononuclear cells;
- IFN,
interferons;
- IgG,
immunoglobulin;
- IL,
interleukins;
- ka,
equilibrium association constant;
- kd,
equilibrium dissociation constant;
- kD,
equilibrium dissociation constant;
- LAG-3,
lymphocyte-activation gene 3;
- MFI,
mean fluorescence intensity;
- MHC-I,
major histocompatibility complex-I;
- MHC-II,
major histocompatibility complex-II;
- NK,
natural killer;
- NSCLC,
non-small cell lung cancer;
- NSG,
NOD scid gamma;
- OD,
optical density;
- PD-1,
programmed cell death-1;
- PD-L1,
programmed cell death ligand-1;
- PD-L2,
programmed cell death ligand-2;
- SPR,
surface plasmon resonance;
- TCR,
T-cell receptor;
- TGF-β,
transforming growth factor-beta;
- TGF-βR,
transforming growth factor-beta receptor;
- TME,
tumour microenvironment;
- TNBC,
triple-negative breast cancer;
- VH,
variable heavy;
- VL,
variable light.
REFERENCES
Cite This Article
TY - JOUR AU - Nerisha Chouhan PY - 2021 DA - 2021/06/28 TI - Bispecific Antibodies for the Treatment of Cancer JO - Materials Highlights SP - 29 EP - 40 VL - 2 IS - 3 SN - 2666-4933 UR - https://doi.org/10.2991/mathi.k.210617.001 DO - https://doi.org/10.2991/mathi.k.210617.001 ID - Chouhan2021 ER -


